EasySep™ Release Human CD19 Positive Selection Kit
Immunomagnetic positive selection kit using particle release technology


概要
The EasySep™ Release Human CD19 Positive Selection Kit is designed to isolate CD19+ cells from fresh or previously frozen peripheral blood mononuclear cells or washed leukapheresis samples by immunomagnetic positive selection.
Desired cells are labeled with antibodies and magnetic particles, and separated without columns using an EasySep™ magnet. Unwanted cells are simply poured off, while desired cells remain in the tube. Then, bound magnetic particles are removed from the EasySep™-isolated CD19+ cells, which are immediately available for downstream applications. Following cell isolation with this EasySep™ Release kit, antibody complexes remain bound to the cell surface and may interact with Brilliant Violet™ antibody conjugates, polyethylene glycol-modified proteins or other chemically related ligands. The CD19 antigen is expressed on mature B cells and B cell progenitors but is lost upon maturation to plasma cells. It is also expressed on follicular dendritic cells.
Desired cells are labeled with antibodies and magnetic particles, and separated without columns using an EasySep™ magnet. Unwanted cells are simply poured off, while desired cells remain in the tube. Then, bound magnetic particles are removed from the EasySep™-isolated CD19+ cells, which are immediately available for downstream applications. Following cell isolation with this EasySep™ Release kit, antibody complexes remain bound to the cell surface and may interact with Brilliant Violet™ antibody conjugates, polyethylene glycol-modified proteins or other chemically related ligands. The CD19 antigen is expressed on mature B cells and B cell progenitors but is lost upon maturation to plasma cells. It is also expressed on follicular dendritic cells.
Advantages
• Highly purified human CD19+ cells isolated in less than 30 minutes
• No-wash removal of EasySep™ Releasable RapidSpheres™
• No-wash removal of EasySep™ Releasable RapidSpheres™
Components
- EasySep™ Release Human CD19 Positive Selection Kit (Catalog #17754)
- EasySep™ Release Human CD19 Positive Selection Cocktail, 1 mL
- EasySep™ Releasable RapidSpheres™ 50201, 1 mL
- EasySep™ Release Buffer (Concentrate), 3 x 1 mL
Magnet Compatibility
• EasySep™ Magnet (Catalog #18000)
• “The Big Easy” EasySep™ Magnet (Catalog #18001)
• EasyPlate™ Magnet (Catalog #18102)
• EasyEights™ Magnet (Catalog #18103)
Subtype
Cell Isolation Kits
Cell Type
B Cells
Species
Human
Sample Source
Leukapheresis, PBMC
Selection Method
Positive
Brand
EasySep
Area of Interest
Immunology
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet 1 | EasySep™ Release Human CD19 Positive Selection Kit | 17754 | 19D102116 or lower | English |
| Product Information Sheet 2 | EasySep™ Release Human CD19 Positive Selection Kit | 17754 | 1000006155 and higher | English |
| Safety Data Sheet 1 | EasySep™ Release Human CD19 Positive Selection Kit | 17754 | All | English |
| Safety Data Sheet 2 | EasySep™ Release Human CD19 Positive Selection Kit | 17754 | All | English |
| Safety Data Sheet 3 | EasySep™ Release Human CD19 Positive Selection Kit | 17754 | All | English |
数据及文献
Data
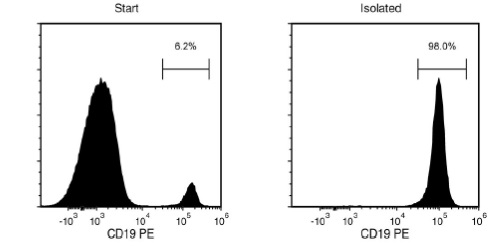
Starting with a single-cell suspension of human PBMCs, the CD19+ cell content of the isolated fraction is typically 97.7 ± 2.3% (mean ± SD using the purple EasySep™ Magnet). In the above example, the purities of the start and final isolated fractions are 6.2% and 98.0%, respectively.
Publications (3)
Science advances 2020 oct
Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche.
Abstract
Abstract
B cell acute lymphoblastic leukemia (B-ALL) blasts hijack the bone marrow (BM) microenvironment to form chemoprotective leukemic BM niches facilitating chemoresistance and, ultimately, disease relapse. However, the ability to dissect these evolving, heterogeneous interactions among distinct B-ALL subtypes and their varying BM niches is limited with current in vivo methods. Here, we demonstrated an in vitro organotypic leukemia-on-a-chip" model to emulate the in vivo B-ALL BM pathology and comparatively studied the spatial and genetic heterogeneity of the BM niche in regulating B-ALL chemotherapy resistance. We revealed the heterogeneous chemoresistance mechanisms across various B-ALL cell lines and patient-derived samples. We showed that the leukemic perivascular endosteal and hematopoietic niche-derived factors maintain B-ALL survival and quiescence (e.g. CXCL12 cytokine signal VCAM-1/OPN adhesive signals and enhanced downstream leukemia-intrinsic NF-$\kappa$B pathway). Furthermore we demonstrated the preclinical use of our model to test niche-cotargeting regimens which may translate to patient-specific therapy screening and response prediction."
Frontiers in immunology 2020
Fli1 Downregulation in Scleroderma Myeloid Cells Has Profibrotic and Proinflammatory Effects.
Abstract
Abstract
Scleroderma (SSc) is an autoimmune connective tissue disease characterized by immune dysregulation, vasculopathy, and fibrosis. We have previously demonstrated that low Fli1 expression in SSc fibroblasts and endothelial cells plays an important role in SSc pathogenesis. Cells of myeloid and lymphoid origin also express Fli1 and are dysregulated in patients with SSc, playing key roles in disease pathogenesis. However, the role for immune Fli1 in SSc is not yet clear. Our aim was to elucidate whether Fli1 contributes to the immune dysregulation seen in SSc. Comparison of the expression of Fli1 in monocytes, B- and T-cell fractions of PBMCs isolated from SSc patients and healthy controls (HC), showed an increase in Fli1 levels in monocytes. We used siRNA transfected human myeloid cells and mouse peritoneal macrophages obtained from Fli1 flox/flox LysMCre+/+ mice, and found that markers of alternative macrophage activation were increased with Fli1 deletion. Coculture of Fli1-deficient myeloid cells and primary human or mouse fibroblasts resulted in a potent induction of collagen type I, independent of TGF$\beta$ upregulation. We next analyzed global gene expression profile in response to Fli1 downregulation, to gain further insight into the molecular mechanisms of this process and to identify differentially expressed genes in myeloid cells. Of relevance to SSc, the top most upregulated pathways were hallmark IFN-$\gamma$ and IFN-$\alpha$ response. Additionally, several genes previously linked to SSc pathogenesis and fibrosis in general were also induced, including CCL2, CCL7, MMP12, and CXCL10. ANKRD1, a profibrotic transcription co-regulator was the top upregulated gene in our array. Our results show that Fli1-deficient myeloid cells share key features with cells from SSc patients, with higher expression of profibrotic markers and activation of interferon responsive genes, thus suggesting that dysregulation of Fli1 in myeloid cells may contribute to SSc pathogenesis.
Acta neuropathologica communications 2017 dec
CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer's PSEN2 N141I neurons.
Abstract
Abstract
Basal forebrain cholinergic neurons (BFCNs) are believed to be one of the first cell types to be affected in all forms of AD, and their dysfunction is clinically correlated with impaired short-term memory formation and retrieval. We present an optimized in vitro protocol to generate human BFCNs from iPSCs, using cell lines from presenilin 2 (PSEN2) mutation carriers and controls. As expected, cell lines harboring the PSEN2 N141I mutation displayed an increase in the A$\beta$42/40 in iPSC-derived BFCNs. Neurons derived from PSEN2 N141I lines generated fewer maximum number of spikes in response to a square depolarizing current injection. The height of the first action potential at rheobase current injection was also significantly decreased in PSEN2 N141I BFCNs. CRISPR/Cas9 correction of the PSEN2 point mutation abolished the electrophysiological deficit, restoring both the maximal number of spikes and spike height to the levels recorded in controls. Increased A$\beta$42/40 was also normalized following CRISPR/Cas-mediated correction of the PSEN2 N141I mutation. The genome editing data confirms the robust consistency of mutation-related changes in A$\beta$42/40 ratio while also showing a PSEN2-mutation-related alteration in electrophysiology.